

Alternatives to Zookeeping
How synthetic biology can replace antibodies and accelerate biomedical research

Recently, I’ve been thinking a lot about antibodies. Antibodies, of course, are remarkable proteins that vertebrate immune systems use to recognize pathogens. But that’s not why I’ve been thinking about them.
In biomedical experiments, scientists often use antibodies to detect proteins of interest. Although antibodies have served well in this purpose for over 70 years of research, they also have quite a few shortcomings. Fortunately, synthetic biology is now capable of providing alternatives to traditional antibodies. These alternatives have the potential to accelerate the progress and improve the reproducibility of biomedical research – but the hardest part may be getting scientists to use them.
How antibodies work
To understand the uses and limitations of antibodies, it’s helpful to focus on a concrete example. Let’s say I want to see if my cells are recombining their chromosomes when I’m activating expression of meiosis-related genes. One way I can do this is through immunofluorescence imaging:
First, I will treat the cells with formaldehyde to crosslink the proteins, and then add a detergent to permeabilize cell membranes. This kills the cells, but stabilizes the proteins and allows the antibodies to diffuse inside during the next step.
Second, I will add a mouse antibody that binds to the human DMC1 protein, which marks chromosomal recombination sites. This antibody is called the primary antibody, since it directly binds the target.
Third, I will wash away the primary antibody, and add a donkey antibody that binds to mouse antibody proteins. This secondary antibody is labeled with a fluorescent dye.1 At this stage, I can also add another dye that labels DNA.

The final binding configuration in an immunofluorescence experiment. Finally, I can wash away the unbound antibody and image the cells using a microscope. If I see fluorescently labeled recombination sites on the DNA of the chromosomes, I’ll be very happy!
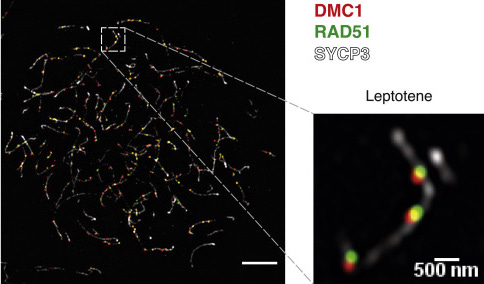
An example immunofluorescence staining for DMC1, RAD51, and SYCP3 on mouse meiotic chromosome, from Hinch et al 2020 (CC-BY)

Besides immunofluorescence imaging, I also use antibodies in a huge variety of other experiments, such as:
Flow cytometry (fluorescent labeling of cell surface markers, allowing cell sorting)
Western blotting (detection of proteins after gel electrophoresis)
Immunoprecipitation (using antibodies to capture a target protein and identify other biomolecules bound to it).2
ELISA (measuring concentrations of biomolecules such as hormones)
But where do these antibodies come from?
To produce antibodies against a particular target biomolecule, scientists will inject an animal with that molecule, typically along with some adjuvant to stimulate an immune response. This process is quite similar to vaccination. The easiest way to isolate antibodies is to purify them from the blood serum of the immunized animal using a process called affinity chromatography.3 This results in a preparation of polyclonal antibody. The name reflects the fact that the preparation is a mixture of antibodies produced by different B cells of the animal.
Alternatively, scientists can isolate individual antibody-producing B-cells, and immortalize them (usually by fusing them with a cancer cell) to produce cell lines that secrete antibodies. The antibodies can then be purified from the cell culture. These antibody preparations are called monoclonal since they contain only a single antibody protein, grown from a single B cell clone.
Usually, this is all done by companies specializing in antibody production. The companies will then sell the antibodies to scientists like me, making a substantial profit.
Why is there a zoo in my freezer?
Immunofluorescence imaging is a powerful technique to see where proteins are localized within cells. However, some complications arise when trying to image multiple proteins at once. The main issue is that you can’t use two primary antibodies produced in the same species, because your secondary antibody won’t be able to distinguish them. You also can’t use a primary antibody from the same species as your secondary antibody, since in that case your secondary antibody would bind to itself (and the animal producing it would have an autoimmune disease).4
Therefore, planning an immunofluorescence experiment is like doing a complicated puzzle. “Hmm, I could stain for Protein #2, except it looks like the only good antibody against it is a mouse monoclonal, and I’m already using one of those for Protein #1. There’s a goat polyclonal from a sketchy supplier but I really don’t trust that. But, if I switch to a rabbit antibody for #1, then I could use the mouse one for #2. And great, I just found a nice-looking chicken antibody for protein #3!”
Now you might understand how I ended up with a freezer box containing antibodies from nearly as many species as a Chinese zodiac!5

Other problems with antibodies
Species compatibility is far from the only problem with antibodies. There are quite a few more. I’ll divide this discussion into two categories: first, problems that are intrinsic to antibodies themselves, and second, problems with how the biomedical research community produces and uses them.
Intrinsic problems
Antibodies can’t diffuse across cell membranes, so intracellular labeling requires permeabilizing the cells (which kills them)
Many cells have Fc receptors, which bind to antibodies. This can cause off-target labeling, although it can be mitigated by adding excess unlabeled antibodies.
It’s very hard to use antibodies quantitatively, especially when using a two-stage staining system. You can see that a protein is there, but you can’t count the molecules.6
As I mentioned above, there are species compatibility limitations.
Extrinsic problems
Antibodies are expensive. Companies charge as much as they can get away with – typically $300 to $650 per vial. If you want to reproduce someone else’s paper that uses 4 antibodies, you can expect to shell out around $2,000.
Many antibody companies are scummy and sell antibodies that don't work! Either not labeling the target, or worse, labeling incorrect things. Some companies are better than others.7 Of the 29 primary antibodies I’ve used for immunofluorescence so far over the course of my PhD, 4 are complete failures, and another 5 are mediocre. And this is mostly using antibodies that were cited in previously published literature! This wastes money, both on buying antibodies that don’t work, and more seriously, on expensive irreproducible experiments. In my field, there was a notorious claim of “adult ovarian stem cells” that wasted years of researcher time, and was largely due to bad antibody labeling.
There are batch-to-batch differences in antibodies, which hurts reproducibility. This is especially bad for polyclonal antibodies, since the proportion of each component of the mixture is not stable over time (and will change radically when switching to a different animal, since no two animals will produce the same antibodies, even if they are genetically identical).
Antibody companies keep most of the information about their antibodies as trade secrets. This is just another example of proprietary bullshit in biomedicine. For example, most companies don’t disclose which part of the protein (i.e., epitope) their antibody binds to.8 Some epitopes are only accessible when the protein is denatured, meaning the antibody will work for things like Western blotting but not immunofluorescence. Other times, epitopes may be present on certain isoforms of the protein but not others. And if you make a mutation to a certain part of the protein for an experiment, you can’t be sure that the antibody will still bind to it. But often there’s no way to know this in advance – the company would rather you buy their antibody and see for yourself. Again, this hurts reproducibility, especially when companies discontinue antibodies or go out of business entirely. Antibodies are definitely patentable, so I wish the companies would patent them (and disclose information) instead of keeping everything as trade secrets.
Researchers often don’t characterize their antibodies by performing the proper controls.9
Antibody production is bad for animals. Santa Cruz Biotech got in serious trouble for this.
How bad are these problems?
Pretty bad. A 2015 article in the news section of Nature had several stories of research projects ruined (and millions of dollars wasted) due to bad antibodies. A companion article estimated the total money wasted to be $350 million/year, as of 2015. A review article in 2016 listed a long series of problems with antibodies in research, and summarized:
Urgent action is required. First, the reproducibility crisis of science, in general, reached an inacceptable level. Second, the huge amount of waste of time and financial resources is not acceptable in a purely economic sense. Third, the use of antibodies of doubtful quality might harm patients and other people dependent on reliable results of diagnostic and other analytical tests. Fourth, bad antibodies and immunoassays damage the reputation of a whole analytical field and on the long term destroys the economic basis of many companies [. . .] And finally, bad antibodies also cause a lot of frustration, which makes the work with antibodies a daunting experience for many scientists.
Unfortunately, not much has changed since then.10 Researchers are still buying and using crappy antibodies. In August 2023, a preprint reported the results of testing 614 commercial antibodies against 65 neuroscience-related proteins. The main conclusions were:
i) more than 50% of all antibodies failed in one or more tests, ii) yet, ∼50-75% of the protein set was covered by at least one high-performing antibody, depending on application, suggesting that coverage of human proteins by commercial antibodies is significant; and iii) recombinant antibodies performed better than monoclonal or polyclonal antibodies. The hundreds of underperforming antibodies identified in this study were found to have been used in a large number of published articles, which should raise alarm.
Synthetic biology to the rescue
Fortunately, synthetic biology can now resolve these issues. First, better methods of producing antibodies can overcome the extrinsic problems. Antibodies are a particular kind of protein, and over the years, scientists have gotten very good at producing antibodies using standardized protein expression methods. This method, called “recombinant antibody production”, requires only the DNA sequence encoding the particular antibody. Basically, this is a better way to make monoclonal antibodies, and the antibody species can be switched at will. However, it is very difficult to make full-size antibodies in microbial expression systems,11 so typically a smaller part of the whole antibody protein is produced, which also gets around the Fc receptor issue.
Nanobodies, which are tiny antibody-like proteins produced by camels and related species, can also be produced recombinantly. Unlike antibodies, nanobodies are single-domain proteins with easy folding characteristics, which makes it much easier to produce them in microbial expression systems. These nanobodies could be linked to other protein binding domains, which would provide a primary/secondary labeling system that would avoid the species compatibility problems and allow me to clean out my freezer.

Recombinant antibody production is already widely used in the pharmaceutical industry, producing blockbuster drugs such as, well, anything ending in -zumab or -ximab. Antibody companies have also started to use recombinant production, although unfortunately they do not disclose the DNA sequences. Finally, the nonprofit plasmid repository Addgene has begun providing a catalog of antibodies with known DNA sequences.
Related technology can also be used to develop new antibodies without using animals. Instead of immunizing an animal, a diverse library of randomized antibody sequences is introduced into bacteriophages.12 Each phage will express a unique antibody on its surface. Next, the desired target molecule is linked to a solid substrate. The phage population is added, and washed until only the ones with the highest binding affinity remain. Subsequently, the target (and phage) is detached from the surface, typically by enzymatic digestion, and the phage is allowed to replicate in bacteria. The process can be repeated, with increasing stringency of washing, to isolate antibodies with extremely high affinity. Although this method selects for high on-target binding, it doesn’t rule out off-target binding,13 so this must be tested in follow-up experiments. A recent (August 2023) preprint reported a way of performing massively parallel protein interaction screens in yeast cells, and similar methods could be used to quickly test specificity against a proteome-scale library.
The ”holy grail” of antibody technology is designing antibodies purely computationally, based on the known target structure. This has been quite challenging, but over the last few years, advances in protein design and machine learning have now made it possible to accurately compute the structures of antibodies based on their sequences. Recently, a preprint released by the small biotech company Absci reported designing 400,000 antibodies against the HER2 protein, of which 421 bound strongly enough to characterize. Although this success rate may seem low, it’s actually quite good compared to previous methods. Using protein structure prediction in combination with more traditional screening methods can also yield great improvements. I anticipate that there will be rapid advances in this area, given its importance to the pharmaceutical industry.
But let’s take a step back and ask: do we really need antibodies? For most research applications,14 what we actually need is a way to specifically label a particular biomolecule. Antibodies are good for this, but de novo designed proteins could be even better, having advantages in affinity, specificity, stability, size, and many other properties. Recently, David Baker’s lab (of Rosetta fame) has developed a method to design completely artificial proteins that can bind any protein target. Although the success rate is still not very high (1 to 17 per 20,000 designs, and the affinity is lower than most antibodies), previously this was completely impossible. Aptamers, which are nucleic acids that bind to target molecules, are another potential option to replace antibodies. I’m optimistic that we may be able to completely replace antibodies for most experimental applications in the future.
How can we move forward?
These technologies are great, but they won’t do any good unless researchers use them. In fact, things like phage display and recombinant antibodies are 30 years old by now, and have still seen limited adoption outside of pharmaceuticals. So, how can we change this?
One option is bottom-up change: individual researchers deciding that it’s worth it to use improved technology. This has the advantage that no coordination is needed, but suffers two major drawbacks. First, being an early adopter of new technology is risky. Things might not work perfectly the first time, and peer reviewers may question why you’re not using standard methods. Second, it’s a lot of work to perform protein expression and purification, and many researchers who specialize in other areas do not have the required skills. This is why antibody companies have a big market. However, those antibody companies won’t switch to new technology unless they know that enough researchers will buy their new products. Although I’d love to use recombinant antibodies for everything, I don’t want to spend time making them instead of researching meiosis, and it’s still rare that manufacturers sell them.
Therefore, top-down change may be more effective. Funding agencies such as the NIH have the potential for a big impact here, because they provide most of the money that goes towards buying antibodies. Here’s an example of how the NIH could set deadlines for phasing out old technology:
Starting in 2026, NIH funds may no longer be used to buy polyclonal antibodies.
Starting in 2028, NIH funds may no longer be used to buy antibodies where the epitope is proprietary or unknown.
Starting in 2030, NIH funds may no longer be used to buy antibodies that are not produced recombinantly. Protein sequences of antibodies purchased with NIH funds must be publicly available.15
This would give antibody manufacturers time to generate and characterize new antibodies (and file patents to protect their IP), and researchers time to transition. Additionally, funding agencies could award grants for developing improved antibody technology, or alternatives to antibodies. By withdrawing funding for old technology and incentivizing adoption of new technology, funding agencies could greatly benefit the biomedical research community – and make it a little less like a circus.
I thank Devon Stork for feedback on this article.
You might be wondering, why not label the primary antibody with the dye and not have to bother with the secondary? In fact, this is also an option, and standard practice for certain methods such as flow cytometry. However, there are two main reasons to use a labeled secondary. First, labeling is expensive, and with one labeled secondary antibody, you can use it in multiple different experiments with different unlabeled primary antibodies. Second, multiple secondary antibody proteins can bind at the same time to one primary antibody protein, and this amplifies the signal.
Regarding the dye, typically this is covalently linked to lysine sidechains on the antibody. Usually, each antibody has more than one dye molecule linked to it, at semi-random sites.
Antibodies targeting DNA-binding proteins can be used for ChIP-Seq, a variant of this method.
This can be performed by immobilizing the antigen of interest on a chromatography resin, to isolate any antibodies that bind to it. However, many manufacturers instead use bacterial proteins that will bind to any antibody, to avoid the cost of preparing a custom chromatography resin. Such preparations of polyclonal antibodies contain all the antibodies present in the host animal’s serum, only a small fraction of which will bind the target. For more information see here and contrast “Antigen-specific affinity” with “Class-specific affinity.”
Interestingly, one antibody I use to label human centromeres comes from a human with an autoimmune disease who produces anti-centromere antibodies.
Mouse, rabbit, rat, goat, chicken, sheep, donkey, human. Unfortunately, I don’t have any dragon antibodies.
Methods like ELISA get around this by having a standard with a known concentration, but this is very hard for Western blotting and almost impossible for immunofluorescence. Part of the reason is that antibodies have a wide range of binding affinities and the binding is complex with the two binding sites.
Santa Cruz is notoriously bad.
In many cases this is because the companies haven’t sufficiently characterized their antibodies and don’t actually know the epitope.
At minimum, they should perform a negative control on samples not expected to contain the target, and show that there is no binding. Ideally, researchers would also do a positive control, and a “blocking control” (positive control plus an excess of the epitope molecule, such that the excess epitope will compete with the target for antibody binding, thus blocking it). Of course, a blocking control is not possible unless the researcher knows the epitope.
The one real improvement has been that most journals now require authors to provide a unique RRID number identifiying which antibody they used. Previously, you might read a paper and not even be able to order the same antibody for a replication experiment, since the authors didn’t provide enough information to unambiguously identify it!
This is because they are multi-part proteins linked by disulfide bonds, and folding them correctly requires very specific conditions. Glycoslyation is also an issue, but this matters more for therapeutic antibodies than for research antibodies. However, in the last decade, some progress has been made on antibody production in engineered E. coli bacteria and Pichia pastoris yeast. Most therapeutic antibodies are produced in hamster cell lines but this is more expensive.
This is called “phage display”, for a more detailed summary see here. Alternatively, yeast cells can be used for yeast display; a eukaryotic expression system allows better antibody folding, but this is lower throughput. Yet another alternative is mRNA display, which links proteins to their mRNAs.
This problem has actually killed people: in a clinical trial, an T-cell receptor targeting a cancer antigen, which was engineered using phage display, actually had off-target binding against a heart protein.
Antibodies produced in vivo don’t have this problem as much, since they are selected against off-target binding through immune central tolerance. Of course, this only applies to antigens from the same species as the species producing the antibody.
The main exception being immunology.
While doing research for this post, I came across a very similar proposal from 2015. Unfortunately, this hasn’t been implemented.
Great articles makes we want to do some bio hacking.